
If you put this question to medical students in prize exams, the bright ones will look puzzled and then give you an answer that goes something like this, heart failure is 'a situation in which the heart fails to generate a sufficient cardiac output to meet tissue requirements'. This is completely wrong!
Heart failure is difficult to define. Here is our attempt to summarise succinctly the main features of the condition.
In the clinical syndrome of heart failure, cardiac output and tissue perfusion are maintained at normal levels at the expense of increased cardiac filling pressures. Increased filling pressures are achieved by activation of neurohormonal compensatory mechanisms. It is the activation of these mechanisms which results in many of the symptoms and signs associated with this condition. Also, the activation of compensatory mechanisms ultimately result in a vicious cycle producing long-term deterioration in cardiac function.
Many common disease processes can damage heart muscle or, lead to problems with the heart valves, impairing the hearts ability to pump blood to the tissues. This may occur acutely in conditions which rapidly destroy or damage significant amounts of myocardium or valvular tissue, for example, large myocardial infarctions, extensive myocarditis, or bacterial endocarditis. More commonly, cardiac dysfunction develops gradually over a period of years due to chronic damage secondary to conditions such as coronary artery disease, systemic hypertension or degenerative valve disease. The unifying feature in these different acute and chronic scenarios is reduction in the efficiency of the heart as a pump.
Within limits, the body will not allow an inefficient heart to compromise tissue perfusion. In the presence of declining cardiac function from whatever cause, neural and hormonal compensatory mechanisms are activated which boost cardiac output and maintain perfusion of the tissues at normal levels. The neural mechanisms involved are increased activity in the sympathetic nervous system, while hormonal compensatory mechanisms centre on the release of renin from the kidney and ADH from the pituitary gland. Activation of these compensatory mechanisms alters the hemodynamics of the entire cardiovascular system raising atrial pressure on the right and left side of the heart and consequently, initially at least, boosting cardiac output. This effect is in accordance with the Frank-Starling relationship. Increasing the filling pressures of the ventricles increases cardiac contractility and output. In some cases these effects may result in a completely compensated state devoid of symptoms, a situation which may persist for years.
However, there is no such thing as a free lunch. Retention of fluid secondary to activation of the renin-angiotensin-aldosterone axis and ADH secretion will indeed elevate atrial pressure by increasing intravascular fluid volume. However, this fluid retention may result in some of the more distressing symptoms of the heart failure syndrome such as peripheral oedema. Elevation of left atrial pressure may result in an increase in hydrostatic pressure in the blood vessels of the lungs with development of pulmonary oedema. Activation of the sympathetic nervous system may boost cardiac output by raising contractility and/or heart rate but sustained activation of sympathetic inputs to the heart damages ventricular myocardium This ultimately results in anatomical re-modelling of the ventricular chambers. This latter effect is important as after an index event (eg a myocardial infarction), even if the damaging pathology is not sustained, the progression of heart failure may be self-perpetuating. Once established, the heart failure syndrome may be relentlessly progressive with worsening symptoms for the patient.
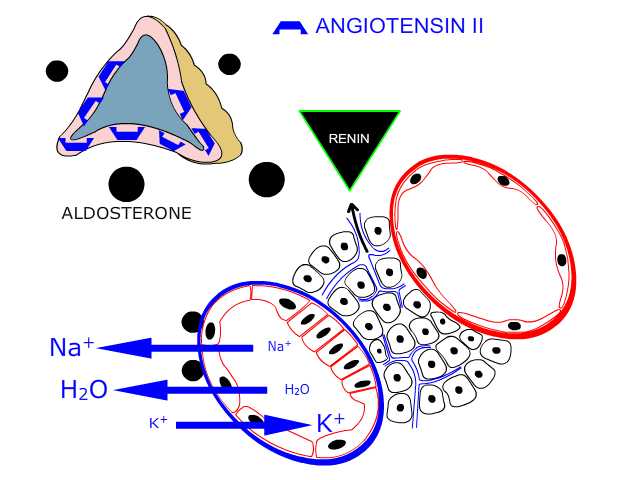
Activation of the renin-angiotensin-aldosterone axis play a key role in generating many of the clinical features of the heart failure syndrome
It is important to realise that activation of the compensatory mechanisms produces many of the physical signs which help us to diagnose the condition. Definitive diagnosis, however, depends on demonstration of elevated left sided cardiac pressures on invasive diagnostic testing.
DisclaimerPrivacy PolicyTerms of UseData Deletion© Acadoodle 2026